Childhood Cancer Awareness Month
September 2024
September is Childhood Cancer Awareness Month, which is a time to shine a light on the thousands of children and their families across the world who are dealing with a cancer diagnosis.
We would love your help to raise awareness and funds this September for the tamariki and whānau who are impacted by childhood cancer in Aotearoa. Keep reading to find out all the ways you can make a difference this Childhood Cancer Awareness Month!
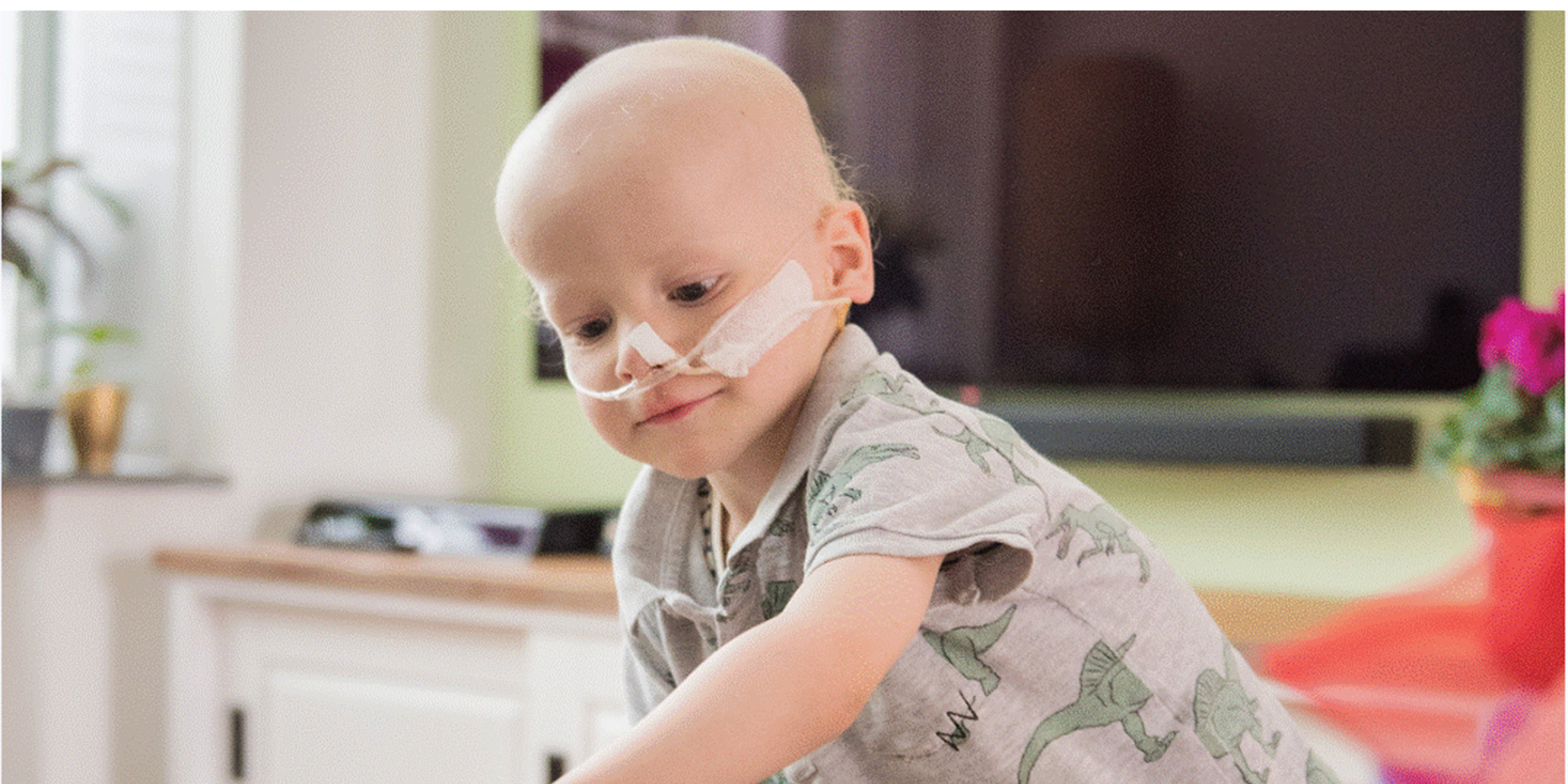
Meet a family impacted by childhood cancer
Isla's story
"We got rushed back in and the doctor told us that Isla’s situation had worsened and that there was a chance she might not wake up again. It was the worst day of our lives." - Keren, whose daughter Isla was diagnosed with cancer at three years old.
How you can get involved this Childhood Cancer Awareness Month
Whether you help by making a donation, raising vital awareness or educating yourself and your whānau on the impact of childhood cancer, you have the power to make a real difference for tamariki with cancer and their families.

Go gold for childhood cancer
The international symbol for childhood cancer is the gold ribbon. This Childhood Cancer Awareness Month, we are asking you to show your support and spread awareness for childhood cancer by using our custom 'Go Gold' filters and GIFs on Instagram throughout September. Just search Child Cancer Foundation for the range.
Create a story
Lachie's Laces
Lachie’s Laces are vibrant yellow shoelaces that have been created in memory of Lachie Sutherland, a bright and beloved boy who died from cancer at just 12 years old in 2020.
Lachie's family and friends wanted to mark what would have been his 14th birthday on 25 September by raising awareness and funds for kids living with cancer in New Zealand.
You can buy Lachie's laces through the link below – all proceeds go to Child Cancer Foundation.

Understanding childhood cancer
We want to help Kiwis learn more about childhood cancer and understand its impact on tamariki and whānau in New Zealand this September. Click 'Read More' to find some great educational articles from our partners at the National Child Cancer Network.
Read more
Different types of childhood cancer
There are many different types of cancer that affect children, some occurring more often than others. Click 'Read More' to learn about the most common types.
Read moreChildhood cancer glossary
There are many complex terms used to describe different aspects of childhood cancer. This glossary explains some of the most common terms that doctors and other health professionals may use.
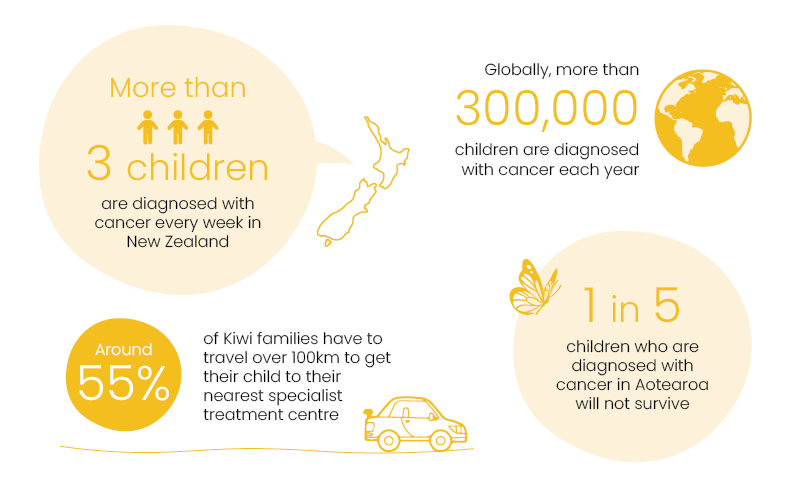
Read nowNew Zealand Children’s Cancer Registry Snapshot 2021
The New Zealand Children’s Cancer Registry (NZCCR) Snapshot is a fantastic visual report which highlights some of the key statistics concerning children diagnosed with cancer during 2021.
More detail on the NZCCR, and previous snapshot reports can be viewed at childcancernetwork.org.nz/nzccr-overview.
Please note that our timeframe for collecting is different from that used in the NZCCR Snapshot (financial year vs calendar year), so statistics may differ slightly.
How we help
Child Cancer Foundation provides families with personalised support at every stage of their experience with childhood cancer. Hear about it first hand through these powerful stories.








Every dollar counts
Please donate today
To ensure each family living through childhood cancer in New Zealand receives the support they need, we need to raise at least $6 million each year – and we receive no funding from the government. That means we rely on the compassion and generosity of Kiwis like you.
Every donation, no matter how big or small, makes a real difference for tamariki with cancer and their whānau. Here are some examples of how your contribution could help.